Il malato di Talassemia Major o Morbo di Cooley è affetto da una grave anemia cronica sin dai primi mesi di vita.
Il malato di Talassemia Major o Morbo di Cooley è affetto da una grave anemia cronica sin dai primi mesi di vita.
La Talassemia è una malattia molto diffusa nell’area del Mediterraneo: i talassemici in Sicilia, che sono più di 2.000, sono curati presso i vari centri specializzati distribuiti nelle province della Regione.
La terapia attualmente in uso consiste in frequenti trasfusioni di sangue, mediamente ogni 15 giorni. Queste comportano un accumulo di ferro nel cuore, nel fegato e nelle ghiandole endocrine che, per essere eliminato, necessita della somministrazione di un farmaco chiamato deferrossamina, viene iniettato al soggetto talassemico sottocute, a mezzo di una pompa ad infusione lenta, per otto-dieci ore al giorno.
Nonostante i notevoli progressi della terapia convenzionale, non si può ancora parlare di guarigione definitiva dalla Talassemia e dalle emoglobinopatie.
Le terapie: dalla mortalità alla sopravvivenza, verso la guarigione
L’evoluzione della terapia nel trattamento dell’anemia mediterranea ha completamente trasformato nel tempo la storia di questa malattia, portandola da una sventura rapidamente fatale a una condizione compatibile con una lunga sopravvivenza e con una qualità di vita molto buona.
Il percorso del progresso terapeutico non è completo: a breve termine i farmaci migliori, e a medio termine le terapie geniche, sono destinati a migliorare ulteriormente le prospettive di vita di questi pazienti.
La successione di risorse terapeutiche ha segnato a tappe la prognosi della talassemia.
La malattia abbandonata a se stessa
Senza cure, il paziente talassemico perde rapidamente emoglobina e globuli rossi, e il midollo osseo non è in grado di sopperire a questa continua distruzione. Senza globuli rossi ed emoglobina la fornitura di ossigeno a tutti gli organi e i tessuti dell’organismo viene progressivamente a esaurirsi, determinando una condizione incompatibile con la vita.
La malattia è quindi mortale, a distanza di settimane o mesi dalla nascita, in funzione della sua gravità.
Le trasfusioni di sangue
La soluzione più istintiva per rimediare al problema è quella delle trasfusioni di sangue. L’immissione di globuli rossi freschi permette al paziente di avere un sangue circolante adeguato per le esigenze dell’organismo.
Il paziente talassemico esegue mediamente due trasfusioni di sangue al mese e relative visite di controllo. Ovviamente questa procedura costituisce un rimedio transitorio: i nuovi globuli rossi si legano comunque all’emoglobina alterata, prodotta dal midollo osseo, e subiscono invariabilmente il processo di distruzione precoce.
Occorrono quindi continue trasfusioni e per tutta la vita.
La somministrazione di globuli rossi giovani offre qualche vantaggio, come pure l’asportazione della milza, riducendo così l’attività distruttiva sulle cellule del sangue.
Si presenta però il problema dell’accumulo di ferro che si libera dall’emoglobina, passa nel circolo sanguigno e va a depositarsi in vari organi — soprattutto fegato, cuore, pancreas — danneggiandone inesorabilmente la funzione, e limitando la vita di queste persone a dieci-venti anni.
La perdita di funzione cardiaca, in conseguenza dell’accumulo di ferro e della fibrosi, è la principale causa di mortalità nei malati talassemici.
Terapia chelante: i farmaci leganti del ferro
Le trasfusioni creano accumuli di ferro in organi vitali. Una drastica soluzione al problema dei depositi di ferro è venuta (negli anni ’70) da farmaci capaci di legarsi a questo metallo circolante (ferrochelanti) ed eliminarlo con le urine. La desferoxamina è stata per decenni l’unico farmaco che ha consentito un cospicuo prolungamento della vita dei malati talassemici, i quali hanno comunque sempre bisogno di trasfusioni.
Questo miglioramento non è senza disagi. La somministrazione del farmaco avviene sottocute con infusione molto lenta (dodici ore) e va ripetuta cinque-sette volte alla settimana.
Non tutto il ferro viene eliminato: il problema dell’accumulo nel cuore e nel fegato viene quindi spostato nel tempo, costituendo infine la principale causa di mortalità (soprattutto cardiaca).
I nuovi ferrochelanti
A partire dagli inizi del 2000 si sono affiancati alla desferoxamina altre molecole capaci di legarsi al ferro, il deferiprone e il deferasirox, assumibili facilmente per bocca, liberando quindi dalla schiavitù dell’infusione continua.
Questi farmaci (talvolta somministrati in maniera orale) sembrano, inoltre, dotati di un’efficacia chelante maggiore. Il Deferiprone riduce l’accumulo di ferro negli organi, soprattutto nel cuore, riducendo la disfunzione cardiaca di cinque volte dopo sei anni di terapia, a parità di condizioni iniziali. Il Deferasirox è approvato per il trattamento del sovraccarico marziale in diverse anemie trasfusioni dipendenti a partire dai 2 anni di età e per il sovraccarico di ferro nelle sindromi talassemiche non trasfusioni dipendenti a partire dai 10 anni di età.
Il trapianto di midollo
È una tecnica con possibilità limitate. Prevede la distruzione delle cellule del midollo, e la successiva reintegrazione con cellule sane geneticamente simili (donatore compatibile). Il trapianto di midollo è ritenuta una metodica più rischiosa rispetto al rischio di mortalità naturale della malattia stessa, e quindi ormai sempre meno utilizzata.
Nuove speranze per la talassemia: la terapia genica
Nuovi studi, in particolare una sperimentazione multicentrica pubblicata nel mese di aprile 2018, spostano il centro dell’attenzione sulla terapia genica. Si tratta di una terapia che fa in modo che il gene dell’emoglobina malato venga sostituito da un gene sano: quest’ultimo, viene modificato in laboratorio e poi veicolato all’interno delle cellule del midollo.
La sperimentazione ha coinvolto 22 pazienti under 35, alcuni con produzione zero di emoglobina e altri con un’importante anemia in cui c’era una piccola quota di emoglobina prodotta dal corpo: i pazienti beta zero hanno diminuito le trasfusioni del 75%, i pazienti con anemia importante hanno totalmente interrotto la terapia trasfusionale.
Per maggiori informazioni su queste ultime novità riguardanti la terapia genica, guarda la video intervista alla dott.ssa Santina Acuto, dirigente biologo dell’unità di ricerca al Campus di Ematologia “Franco e Piera Cutino”.

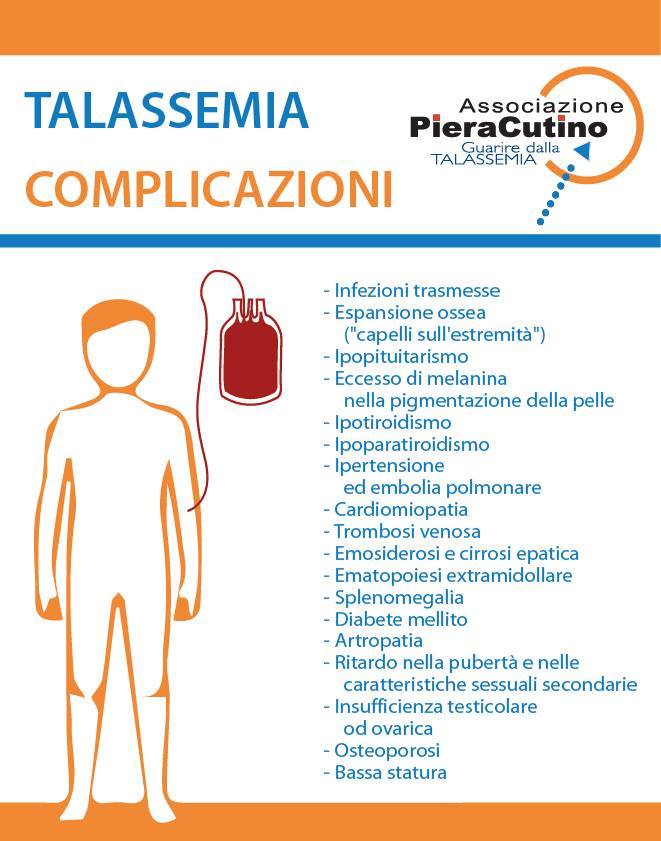
La qualità della vita del paziente talassemico e le complicazioni della talassemia
I pazienti con trasfusioni e chelazioni regolari hanno una buona qualità della vita e possono anche diventare genitori. Mentre fino a trent’anni fa l’aspettativa di vita dei pazienti non superava i 25 anni, oggi la talassemia è una malattia a prognosi aperta!
Tuttavia, le persone affette da Talassemia Major vanno incontro a una serie di complicazioni che derivano dalla malattia e dalle terapie impiegato per la cura dell’anemia mediterranea. Nella scheda vengono elencate alcune delle principali problematiche correlate alla patologia.